Science
Cracking the Dark Genome: What This Indian Scientist And His Team Have Discovered About The 'Greatest Show On The Planet'
Aravindan Neelakandan
Feb 12, 2021, 04:27 PM | Updated 04:27 PM IST
Save & read from anywhere!
Bookmark stories for easy access on any device or the Swarajya app.


If physicists have their dark matter, molecular biologists have their dark DNA or dark genome.
Now a dynamic team of young biologists from across the planet, the Prabakaran Group, is cracking its secrets like never before.
Let's start from the basics.
What is a gene?
The gene is that part of the DNA strand that codes for a protein.
That is a strict and conservative textbook definition. But then, for quite a considerable number of years now, scientists studying the genome have been facing a problem.
The textbook definition of gene that we all learnt in our high school biology is a little inaccurate points out Sudhakaran Prabakaran from the Department of Genetics at the University of Cambridge.
Genes need not code for proteins always. Genes can code for non-coding RNAs too.
There are 58,825 genes, of which 19,968 are protein-coding genes or Open Reading Frames (ORFs).
These ORFs make all the proteins and their isoforms. But this number of protein coding genes, approximately 20,000, has remained remarkably constant over the last 500 million years across all species because they have been conservatively defined, he says.

Have you played those famous puzzles where you have to identify meaningful words in a maze of random letters which are mostly gibberish?
We have a similar situation in the case of most genomes of the organisms.
There are a lot of non-coding DNA and between them there exists the genes or the coding DNA.
Scientists used to call them ‘junk DNA’ in a dismissive manner.
Consider this. In 2001 as the human genome project progressed scientists found that in an average human cell close to 98 percent of the DNA was 'junk DNA' and only 1.5 per cent codes for protein-coding ORFs.
We know that in Darwinian evolution, species arise from a common ancestor and the evolution of life consists of repeated branches.
With common origins come commonalities; like common shape of the organs – even with different functions.
Fore arms of humans, wings of the birds, the flippers of the whales are all examples of such organs.
Evolutionary science calls these organs homologous organs. Similar to homologous organs, we also have homologous proteins. Genes which code for the homologous proteins are homologous genes.
When two species diverge from a common ancestor then the homologous genes have a similar coding sequence. More the divergence and branching the coding sequence will also have more differences.
These are all clearly understood evolutionary science 101.
In 1996, scientists studying yeast genome made an interesting discovery. They found a set of genes which have no homologs in other species. It is almost a forgone conclusion that almost no genes could exist without being homologous.
So, the existence of these genes was initially a shock. These genes are called De Novo genes .
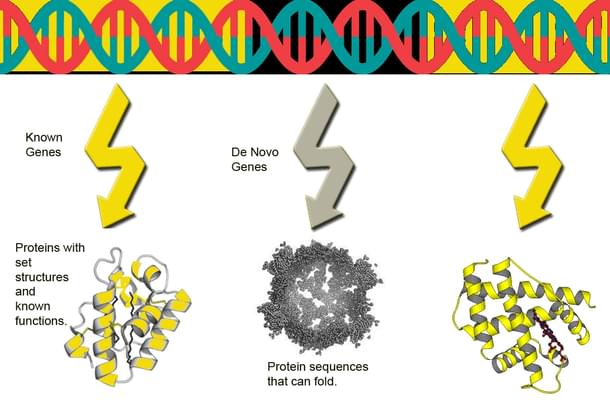
Subsequently, in the sequencing of the genomes of other organisms, researchers discovered more such De Novo genes. These observations that challenged the concept of the gene were dismissed as biological noise and of no consequence.
The non-coding DNA, the novel genes and proteins they code, the processes which drive evolution – all these are pieces of the same puzzle and yet they do not seem to fit.
Then in 2014, while investigating a mouse genome, Prabakaran and colleagues showed that ‘novel’ proteins are not only made pervasively throughout its genome but these novel proteins are biologically regulated too.
Now these puzzles are coming together slowly, very slowly with years of careful research but with astonishing smooth fit.
This is allowing us to see the evolutionary process in action at the level of DNA – also providing humanity knowledge to fight some of the deadliest diseases it has been facing through its history.
Dismissively called ‘junk’ DNA, the non-coding DNA has been getting identified as a place from where new genes arise, the so-called de novo genes. Most of these genes do not survive. But some do and they become part of the genome.
Because these new and as yet unannotated genomic regions that code for these novel proteins are undefined and don’t come under the conventional definition of a protein coding gene, Prabakaran and his group have gathered all such evidence from all over the world, have catalogued and reclassified them (Neville et al).
They call these novel regions as novel Open Reading Frame or n-ORFs.
Now we are arriving at the fundamental question. From the non-coding region between genes, to these becoming expressive and ultimately becoming novel-protein coding genes – how does this transition happen?
Let us now add another piece from yet another puzzle box from biology.
We know that speciation or branching out of a new species from a common ancestor is generally a slow process. It takes quite a lot of geological time.
However, there are also instances where the speciation happens quickly – diverse species from a common ancestor radiate in a very short geological period. Selection forces like change in the environment, food scarcity etc. can force such diversification.
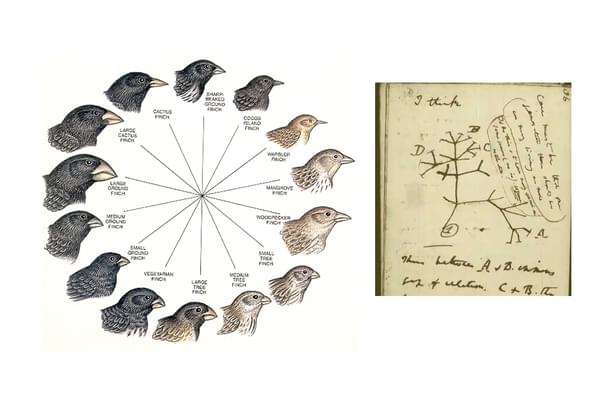
In fact, it was an identification of such diversified varied species of finches that led Darwin to his formulation of natural selection.
Such emergence of such multitude of new forms from an ancestral form because of the changes in the environmental factors is called adaptive radiation.
Now with advancements in genomic and proteomic technologies we are in a position to study this rapid speciation process at the gene-expression level.
In 2020, a team of scientists from the Indian Institute of Science Education and Research Pune, and genetics department of the University of Cambridge studied the underlying molecular processes in the species of the family of Cichlids.
These family of fish exhibit very high species diversity and hence lend themselves to study the enigmatic process of speciation. The geographical hot spot of their species diversity is in the great lakes of east Africa.
Per lake the species diversity for this family of fish is 250 to 500. In Lake Victoria which is part of the East African great lakes, the time taken for this is 100,000 years – almost a couple of eye-blinks time in terms of evolutionary time scale. To put things in perspective, the human-chimpanzee divergence took more than 4 million years.
As the genomes of most of these species of cichlids are remarkably same, ‘the very subtle differences between the genomes of these organisms’ are not enough to explain the manifest differences in the species. But genomic sequencing of five cichlid species has suggested the possibility of the divergence arising from the actions of novel ORF.
An earlier study by Dr. Prabakaran and the team has shown the non-coding genome to be very active across species even though they are not considered as protein-coding genes.
So, what if adaptive radiation involves the novel-ORF component of the genome?
Along with proteins and DNA the messenger in between is the RNA. It plays the obviously important role in the final product – the protein. What it leaves out in translation from DNA source code is called intron.
That intron is also part of the non-coding DNA. Studying of this RNA complex of an organism is called transcriptomics. Combined study of the organism which includes genomics, proteomics and transcriptomics is called Proteogenomics.
Now the team from Pune and Cambridge employed Proteogenomic study and evolutionary analysis of two species of Cichlid fish to test their hypothesis.
The Proteogenomic analysis was done on two cichlid species: one (Pundamilia nyererei) is a rock-dwelling lacustrine fish which is also carnivore with yellow flanks and red dorsal regions - the last trait associated with sexual selection.
The other (Oreochromis niloticus) is a river-dwelling fish which is also an omnivore with a primarily plant-based diet and has plain colouration. Both fishes are genetically quite similar but vary very much in their physical appearance.
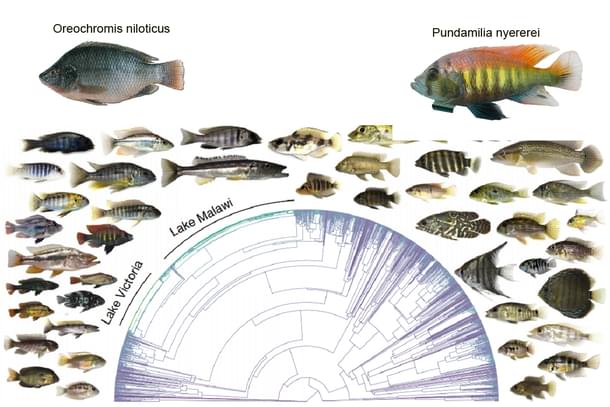
The study compared, between the two species, the expression of transcriptions made from the nORF in two metabolically-active tissues, the testes and liver; liver because the diet distinctions between the species would make for the divergence seen in liver transcriptomes and testes because that is where the highest expression levels of de novo genes have been observed.
The results identified eight novel-nORFs and the divergence in the n-ORF functional products in the two species.
Making these eight n-ORF as data sets along with a control, the branching of these two species in the larger tree of evolution was then analysed to see if it can reveal the divergence time. This involves a software called BEAST(v1.10.4), a cute acronym of what it does: it runs Bayesian Evolutionary Analysis Sampling Tree model.
So, what divergence we obtained through the Proteogenomic study of the liver and testes samples of the two species was entered into a molecular clock mechanism specifically set based on the fossil records of cichlid evolutionary history - 45.5 million years ago (MYA) with a standard deviation of 0.5 MYA for the complete group of cichlids.
Of the eight, four n-ORF show divergence considerably greater than 40 million years, which is more than the age of the oldest cichlid fossils, meaning they most probably did not contribute to the speciation of cichlids. The remaining four n-ORF show a divergence time that vary from 3 to 38 MYA.
This, opine the researchers, strongly suggest the role of nORF in that important evolutionary process- speciation.
Any species can be in the place of Darwin’s finches or cichlids – that is with the environmental changes – food scarcity to geological events to change in habitat – which exerts extreme selection pressure.
Those species which have the ‘evolvable’ genome then undergo adaptive radiation.
This evolvable genome, point out the scientists, are more likely to be in the noncoding regions than in the coding regions. This is because the known genome or the coding genome components have already evolved and got fixed.
The non-coding genome or the dark genome is still in a sort of flux – kind of like molecular biology equivalent of non-collapsed wave function – containing possibilities that may help the organism adapt to the selection pressures.
The team has not only been working on cichlids. And the value of studying n-ORF components of the genome go beyond the discovery of the evolutionary processes that have shaped our past.
In January of 2021, two works have been published by the same Indian and UK team of geneticists on n-ORF. They are of extraordinary exploratory value.
In one of these papers (Neville et al) they cataloged, curated, annotated, and released ~194,000 n-ORFs in the human genome. This is an important addition to the growing n-ORF database.
Their annotation of the nORF sequences for disease mutations has the potential to look into new and novel ways of fighting genetic diseases, rare and prevalent.
The other paper (Erady et al) showed that 'nORFs proteins can form structures, can undergo biochemical regulation like known proteins, and be targeted by drugs in case they are disrupted in diseases. This study essentially underscored the fact that these novel proteins cannot be dismissed as biological noise but are made for specific purpose that could be compromised in diseases.
![Malarial parasite Plasmodium falciparum has two stages in life cycle, with different gene exprssions: [Illustration from: David Fidock et al 2014]](https://swarajya.gumlet.io/swarajya/2021-02/e47d2d6d-575e-4dd7-94ca-f47a11b42454/mp.jpg?w=610&q=75&compress=true&format=auto)
This February Malaria Journal published another research finding by Gunnarsson & Prabakaran.
Malaria caused by Plasmodium falciparum remains to this day the deadliest of the infectious diseases that humanity is fighting with. Even with the only licensed vaccine, the protection is limited and what is more, the resistance to the drug is increasing.
The genomic study of this malarial parasite has been done through conventional ways.
Now Gunnarsson and Prabakaran have made a proteogenomics analysis.
The parasite has stages in its life cycle where it infects the mosquitoe (oocyst sporozoite) and humans (salivary gland sporozoite). The transformation from one stage to another involves 'drastic changes in gene expressions' - which genes to be switched on for which proteins and which genes to be switched off.
Through proteogenomics analysis nORFs have been reported now in the parasite. This research, right now computational and exploratory, can help us create innovative means to fight this deadly organism.
All these findings change the way we look at the mainstream genomic DNA and the orphans or novel-ORF DNA sequence. Says Dr Sudhakaran Prabakaran:
All these observations essentially mean that under stress, or physiological changes, or adaptive radiation nature tinkers with the ‘evolvable’ noncoding genome to make new parts as needed. If this is true, we speculate that all the known protein-coding genes in a population or species are essentially playing the role of house-keeping genes and the n-ORFs are the ones that perform specific functions.
In other words, the ‘dark genome’ is actually the one that is responsive to the selection pressures and hence is the inner driver of evolutionary process.
This also changes the way we view the genes. Early on we have viewed genes more like static beads on a string. We all can remember how we learned Mendel’s laws of genetics where justifiably we visualised genes as kind of static particles.
Then came Barbara McClintock. She showed that the genes were not static but they are dynamic and move within the genome, controlling expression of other genes.
As the Prabhakaran team looks into the secrets of the dark genome, they are uncovering the processes which run the ‘greatest show on the planet’ - evolution and making us explore novel ways of fighting the diseases.
Dr. Sudhakaran Prabakaran points out that the results of these separate studied cutting across the species show that it validates our hypothesis that ‘we have to understand the ‘dark genome’ in order to better understand biology.’
Journal References for this piece:
Puntambekar, S. et al., 2020. Evolutionary divergence of novel open reading frames in cichlids speciation. Scientific reports, 10(1), p.21570.
Neville, M.D.C. et al., 2021. A platform for curated products from novel open reading frames prompts reinterpretation of disease variants. Genome research. online: http://dx.doi.org/10.1101/gr.263202.120.
Erady, C. et al., 2021. Pan-cancer analysis of transcripts encoding novel open-reading frame (nORFs) and their potential biological functions. NPJ genomic medicine, 6(1), p.4.
Gunnarsson, S., Prabakaran, S. In silico identification of novel open reading frames in Plasmodium falciparum oocyte and salivary gland sporozoites using proteogenomics framework. Malaria Journal, 20, 71 (2021). https://doi.org/10.1186/s12936-021-03598-1
My personal gratitude to Dr. Prabhakaran for the inputs and his valuable time which made this article possible.
Aravindan is a contributing editor at Swarajya.
Get Swarajya in your inbox.
Magazine





